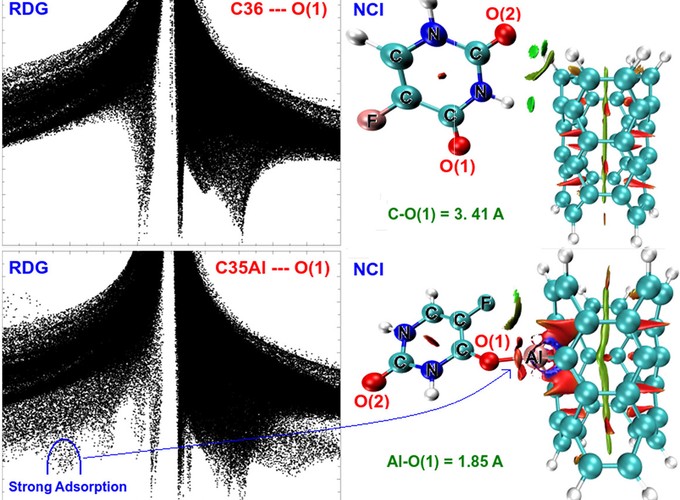
Density functional theory (DFT) is used to examine the formation possibility of a stable interaction between 5-fluorouracil (5-FU) drug molecule and a pristine, boron (B), aluminum (Al), and gallium (Ga)-doped carbon nanotube (CNT). The structural, electronic, optical and reactivity properties of mentioned complexes are investigated in detail. Adsorption energies between the CNT and 5-FU are calculated in the range of −3.79 and −4.38 kcal/mol. Herein, the adsorption of the 5-FU on B-doped CNT is very weak, while stronger adsorption takes place in the case of Al- and Ga-doped CNTs. The results mean that the Al and Ga dopant increases the adsorption capacity of CNT with enhancing its interactions with oxygen atoms of the 5-FU. The charge transfer from adsorbed the 5-FU to Al- and Ga-doped CNTs was confirmed by the natural bond orbital, Mulliken charges, FBO and LBO analyses. It is found that the adsorption of 5-FU on Al-doped CNT is relatively stronger than that of Ga-doped CNT. The NCI-RDG analyses also verify these findings. The first absorption peaks suggest that the B-, A, Ga doped CNTs can absorb in the visible light region. Finally, Al-doped CNT has more desirable properties to use it as a drug delivery system.